
Left Ventricular Hypertrophy as a Marker of Cardiac Involvement in AL Amyloidosis
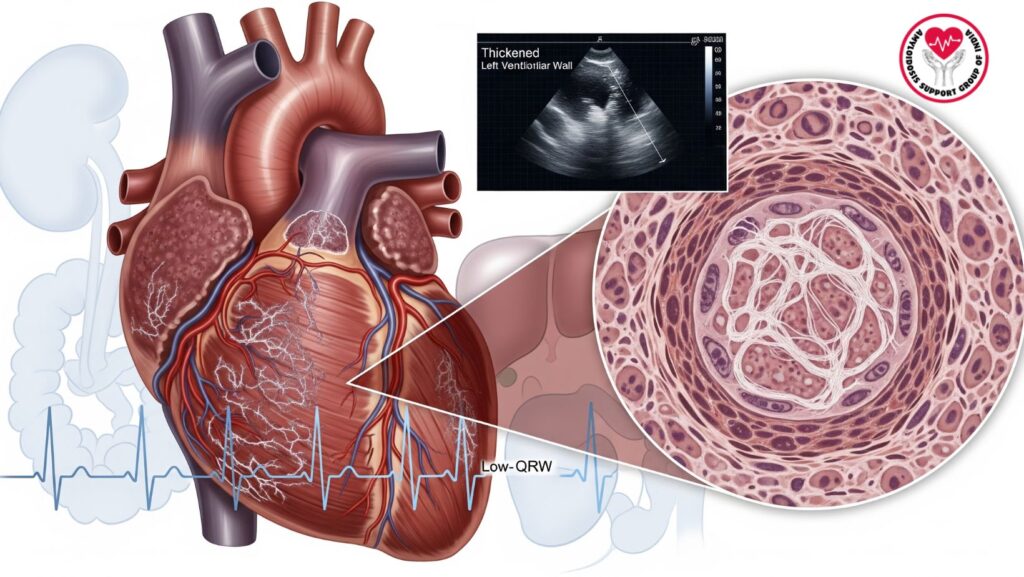
Table of Contents
Introduction
AL (primary) amyloidosis is a systemic illness with deposition of misfolded immunoglobulin light chain amyloid fibrils in multiple organs, including the heart, kidneys, liver, and gastrointestinal tract. Cardiac involvement is a key predictor of morbidity and mortality.
Left ventricular hypertrophy (LVH) on echocardiography is a characteristic finding of cardiac amyloidosis, representing infiltrative myocardial deposition of amyloid fibrils. Awareness of LVH in the setting of systemic AL amyloidosis is important for:
- Early recognition of cardiac involvement
- Prognosis
- Selection of therapy
- Monitoring of therapy response
This complete guide addresses: - Pathophysiology of cardiac involvement in AL amyloidosis
- Diagnostic strategies to LVH in amyloidosis
- Clinical presentation and complications
- Prognostic significance
- Management approaches and therapeutic implications
- Directions for future research on cardiac amyloidosis
Epidemiology of Cardiac Involvement
- Cardiac involvement is present in ~50% of AL amyloidosis patients.
- LVH is among the most frequent echocardiographic findings.
- With increasing older age and greater systemic amyloid burden, there is increased prevalence.
- Male predominance reported in some reports.
Risk Factors for Cardiac Amyloidosis
- Increased circulating free light chains
- Prolonged delay in diagnosis of AL amyloidosis
- Multi-organ involvement, especially renal and hepatic
- Genetic variants that affect amyloidogenic potential
Pathophysiology of Left Ventricular Hypertrophy in AL Amyloidosis
Mechanisms of LVH
- Amyloid infiltration:
- Extracellular deposition of light chain fibrils in myocardial interstitium
- Results in augmented ventricular wall thickness with no actual myocyte hypertrophy
- Myocardial stiffening:
- Impacts diastolic relaxation, leading to diastolic heart failure
- Residual ejection fraction in early phases
- Microvascular involvement:
- Amyloid deposits in coronary arterioles decrease perfusion
- Also leads to ischemia and fibrosis
- Neurohormonal activation:
- Long-term myocardial stress initiates fibrosis and LV remodeling
Pathologic Features
- Symmetric LV hypertrophy
- Interstitial amyloid deposits
- Inflammatory infiltration is minimal
- Sparing of epicardial coronary arteries early on
Clinical Manifestations
#
Early Cardiac Symptoms
- Fatigue and intolerance to exercise
- Mild dyspnea with exertion
- Palpitations or asymptomatic LVH
Advanced Cardiac Symptoms
- Congestive heart failure (diastolic, preserved EF)
- Peripheral edema and ascites
- Pulmonary congestion
- Arrhythmias (atrial fibrillation, conduction blocks)
- Syncope or presyncope
Red Flags for Cardiac Amyloidosis
- LVH on echocardiogram with low-voltage ECG
- Discrepancy between wall thickness and QRS amplitude
- Rapidly progressive heart failure in older adults
Diagnostic Evaluation
Echocardiography
- Imaging modality of first choice for the detection of LVH
- Suggestive findings of cardiac amyloidosis:
- Symmetric wall thickening of the LV
- Biatrial enlargement
- Diastolic dysfunction (restrictive filling pattern)
- Speckled or granular appearance of the myocardium
Electrocardiography (ECG)
- Low-voltage QRS complexes in the presence of LVH
- Pseudoinfarction patterns (Q waves in the absence of CAD)
- Conduction disturbances (bundle branch blocks, AV block)
Cardiac Biomarkers
- NT-proBNP: Raised due to myocardial stress
- Troponin T/I: Suggests chronic myocardial injury
- Light chain ratio: Establishes systemic amyloidosis
Cardiac MRI
- Late gadolinium enhancement demonstrates diffuse subendocardial or transmural deposition
- Measures myocardial involvement
- T1 mapping and extracellular volume fraction measure amyloid burden
Endomyocardial Biopsy
- Definitive gold standard diagnosis
- Establishes amyloid deposition in myocardial tissue
- Congo red staining and mass spectroscopy employed for amyloid typing
Prognostic Implications
- Cardiac involvement is the greatest predictor of mortality for AL amyloidosis
- Median survival:
- In the absence of cardiac involvement: ~4 years
- With cardiac involvement: 6–12 months
- LVH is associated with:
- Greater risk of heart failure
- Higher NT-proBNP and troponin levels
- Reduced response to chemotherapy
Mayo Clinic Staging
- Includes NT-proBNP, troponin, and free light chain difference
- Higher stage predicts poorer prognosis
- LVH frequently parallels advanced stages
Treatment Strategies
General Principles
- Early identification of LVH enables timely initiation of treatment
- Multi-disciplinary management involving hematology, cardiology, nephrology
Disease-Modifying Therapy
- Bortezomib-based regimens: First-line treatment of choice
- Cyclophosphamide + dexamethasone in patients with hepatic or renal impairment
- Autologous stem cell transplant in selected individuals
- Choice of treatment based on degree of cardiac involvement
Heart Failure Management
- Diuretics: Manage congestion, mindful of avoiding hypotension
- Beta-blockers and ACE inhibitors: With caution, usually ill-tolerated
- Pacemakers or ICDs: In arrhythmias or conduction abnormalities
Advanced Interventions
- Heart transplantation in solitary cardiac amyloidosis (exceptional)
- Combined organ transplant (heart and kidney) for multi-organ failure
Case Studies
- Patient X:
- 61-year-old man with LVH, dyspnea, and nephrotic-range proteinuria
- Echocardiogram: Symmetric LV thickening
- Outcome: Poor, died within 6 months despite treatment
- Patient Y:
- Incidental finding of early LVH on echocardiogram
- Aggressive chemotherapy given
- Outcome: Stabilization and better cardiac biomarkers
- Patient Z:
- Advanced LVH with low-voltage ECG and arrhythmias
- Multidisciplinary treatment and palliative management
- Outcome: Symptom control, median survival ~3 months
Clinical Pearls
- AL amyloidosis LVH is infiltrative cardiomyopathy, rather than usual hypertrophy
- Echocardiographic thickening of the LV wall with low-voltage ECG is extremely suggestive
- Early diagnosis profoundly affects choice of therapy and prognosis
- Multi-organ evaluation is essential for wholesome management
- Palliative treatment should be considered in advanced disease with extreme LVH
Future Directions
- New treatments: Monoclonal antibodies that specifically target amyloid fibrils (e.g., CAEL-101)
- Imaging advances: T1 mapping, strain imaging for early detection
- Biomarkers: NT-proBNP and troponin kinetics for monitoring response to therapy
- Genetic studies: Elucidating light chain amyloidogenic variants
- Personalized medicine: Therapy according to severity of LVH and amyloid burden
Conclusion
Left ventricular hypertrophy is a critical indicator of cardiac involvement in AL amyloidosis. Diagnosing LVH in systemic amyloidosis:
- Allows earlier diagnosis
- Informs risk stratification and choice of therapy
- Allows prediction of prognosis and survival
- Facilitates planning for multidisciplinary care
Early identification of LVH can significantly impact patient outcomes by allowing timely intervention and careful monitoring of cardiac function in AL amyloidosis.
