
Key Clinical Insights: Identifying AL Amyloidosis in Unexplained Liver Failure
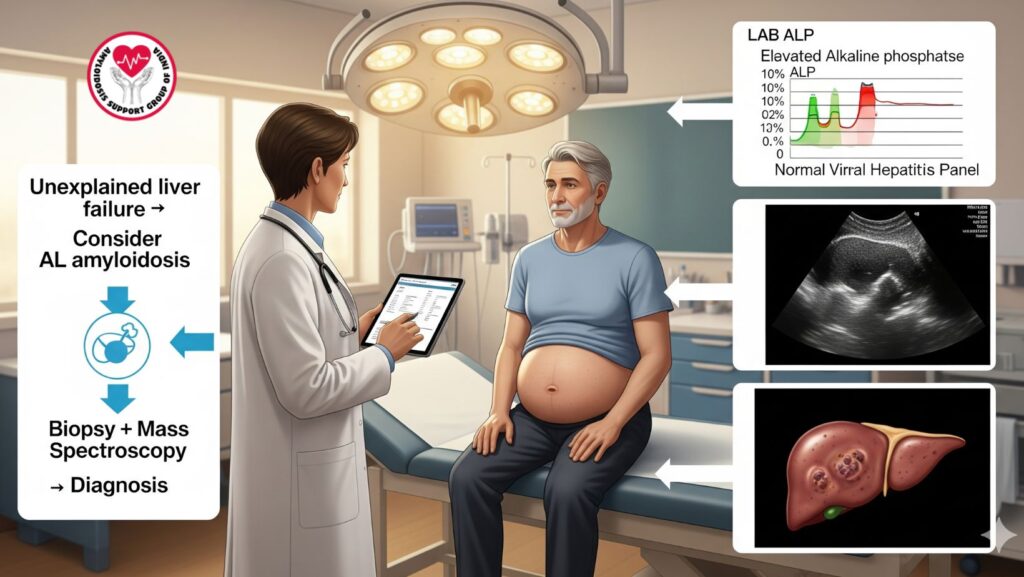
Table of Contents
Introduction
AL amyloidosis is a rare systemic disease due to immunoglobulin light chain deposition in numerous organs, including the liver. Although liver disease can be mild at presentation, unexplained liver failure must raise consideration of AL amyloidosis, particularly when initial testing for viral, autoimmune, or metabolic liver disease is unrevealing.
This article presents salient clinical observations for the identification of AL amyloidosis in patients presenting with unexplained liver failure, with emphasis on diagnostic strategies, laboratory and imaging findings, and prognostic factors.
1. Introduction to AL Amyloidosis and Involvement of the Liver
- Definition: AL (light-chain) amyloidosis results from aberrant immunoglobulin light chains secreted by clonal plasma cells.
- Liver involvement: Seen in \~30–60% of systemic disease; can manifest with:
- Hepatomegaly
- Mild to moderate jaundice
- Alkaline phosphatase (ALP) elevation
- Malaise and fatigue
Key Point: Liver involvement in AL amyloidosis is frequently progressive and refractory to conventional treatment of liver disease.
2. Why Early Recognition Matters
- Prognosis: Liver-involving AL amyloidosis has a poor prognosis, particularly when bilirubin is elevated or synthetic function is impaired.
- Impact of treatment: Early diagnosis enables prompt use of chemotherapy, bortezomib-based treatment, or stem cell transplantation, which can retard organ damage.
- Prevent misdiagnosis: Patients can be treated inappropriately for viral, autoimmune, or drug-induced liver disease, with delayed effective therapy.
3. Clinical Clues Indicative of AL Amyloidosis in Liver Failure
3.1 Patient History
- Unexplained fatigue, weight loss, or anorexia
- History of proteinuria, heart failure, neuropathy, or other systemic involvement
- No pre-existing risk factors for autoimmune or viral liver disease
3.2 Physical Examination
- Hepatomegaly, usually smooth and painless at first
- Later development of jaundice in course of disease
- Edema or ascites secondary to hypoalbuminemia and portal hypertension
3.3 Laboratory Findings
- Differently elevated ALP in relation to AST/ALT
- Slightly elevated bilirubin early on, increasing as disease worsens
- Normal viral hepatitis and autoimmune panels
- Proteinuria can suggest simultaneous renal involvement
- Serum free light chain assay might show abnormal light chain ratio
Key Insight: A combination of hepatomegaly, cholestatic lab pattern, and systemic signs should heighten suspicion.
4. Imaging Clues
- Ultrasound: Hepatomegaly, occasionally nodular liver surface, ascites without splenomegaly
- CT scan: Heterogeneous parenchyma, liver enlargement
- MRI: Variable signal; decreased enhancement, indicative of infiltrative process
- Nuclear imaging: SAP scintigraphy can measure systemic amyloid burden
Clinical Tip: Imaging by itself cannot be used to confirm AL amyloidosis, but may support suspicion in unexplained liver failure.
5. Definitive Diagnosis
5.1 Tissue Biopsy
- Liver biopsy usually needed for confirmation
- Congo red staining: Demonstrates apple-green birefringence under polarized light
5.2 Protein Typing
- Mass spectroscopy determines specific light chain type (lambda or kappa)
- Directs disease-directed therapy
5.3 Other Supportive Diagnostics
- Serum and urine immunofixation: Identifies monoclonal light chains
- Echocardiography: Assesses simultaneous cardiac involvement
- Renal biopsy in the event of proteinuria
Key Insight: Early biopsy and amyloid typing are essential for accurate diagnosis and prognosis.
6. Differential Diagnosis
Physicians should investigate alternative reasons for unexplained liver failure:
- Viral hepatitis (A, B, C, E)
- Autoimmune hepatitis
- Drug-induced liver injury
- Non-alcoholic fatty liver disease (NAFLD)
- Metabolic liver disorders (Wilson’s disease, hemochromatosis)
- Infiltrative diseases (sarcoidosis, lymphoma)
Red Flag: Repeated cholestatic pattern with negative routine tests should raise consideration of AL amyloidosis.
7. Prognostic Implications
- Poor prognosis: Hepatic involvement elevates risk of rapid hepatic failure
- Elevated bilirubin (>2 mg/dL) or hepatic synthetic dysfunction portends poorer outcomes
- Multi-organ involvement (heart, kidneys) decreases survival even further
Key Message: Early diagnosis and initiation of systemic therapy can salvage advanced cases.
8. Clinical Management Principles
8.1 Disease-Directed Therapy
- AL amyloidosis: Cyclophosphamide, dexamethasone, bortezomib-based chemotherapy
- Stem cell transplant: Reserved for eligible patients
- Treatment is intended to decrease light chain production, retard organ damage
8.2 Supportive Care
- Management of ascites and edema (diuretics, sodium restriction)
- Symptom relief and nutritional support (pruritus, fatigue)
- Monitoring of systemic and liver function involvement
Clinical Tip: Supportive care enhances quality of life as systemic therapy targets the underlying amyloidosis.
9. Case Illustration
Patient: 61-year-old man with progressive fatigue and hepatomegaly
Workup:
- Labs: Hyper ALP, viral hepatitis panel is normal
- Imaging: Nodular hepatomegaly, mild ascites
- Biopsy: Congo red positive for amyloid
- Mass spectroscopy: Lambda light chain AL amyloidosis
Management: - Bortezomib-based chemotherapy started
- Supportive care: Diuretics for ascites, nutrition
- Outcome: Improvement of liver function, alleviation of symptoms
Takeaway: Early workup of unexplained liver failure can result in timely AL amyloidosis diagnosis and treatment.
10. Key Clinical Pearls
- High suspicion: Raise AL amyloidosis in unexplained liver failure with cholestatic labs
- Early biopsy: Tissue confirmation and protein typing direct therapy
- Systemic assessment: Assess heart, kidney, and peripheral nervous system
- Prompt intervention: Early treatment prevents organ progression and improves survival
- Supportive care: Key to preserving quality of life with therapy
11. Summary Table: Recognizing AL Amyloidosis in Liver Failure
| Feature | Clinical Significance |
| —————————————- | —————————————- |
| Unexplained cholestatic liver enzymes | Raise suspicion for infiltrative disease |
| Hepatomegaly | Common early sign |
| Normal viral & autoimmune panels | Excludes common liver diseases |
| Systemic signs (proteinuria, neuropathy) | Supports systemic amyloidosis |
| Tissue biopsy + MS typing | Confirms diagnosis and directs therapy |
| Early intervention | Improves prognosis |
12. Conclusion
AL amyloidosis must always be kept in mind in patients with unexplained liver failure. Important messages for clinicians:
- Have high index of suspicion in the case of negative routine evaluations
- Biopsy and mass spectroscopy early on are essential for accurate diagnosis
- Disease-directed treatment along with supportive care leads to better outcomes
- Delayed diagnosis can result in rapid hepatic failure and poor prognosis
