
AL Amyloidosis and Liver Involvement – A Comparison of Survival in Jaundiced vs. Non-Jaundiced Patients
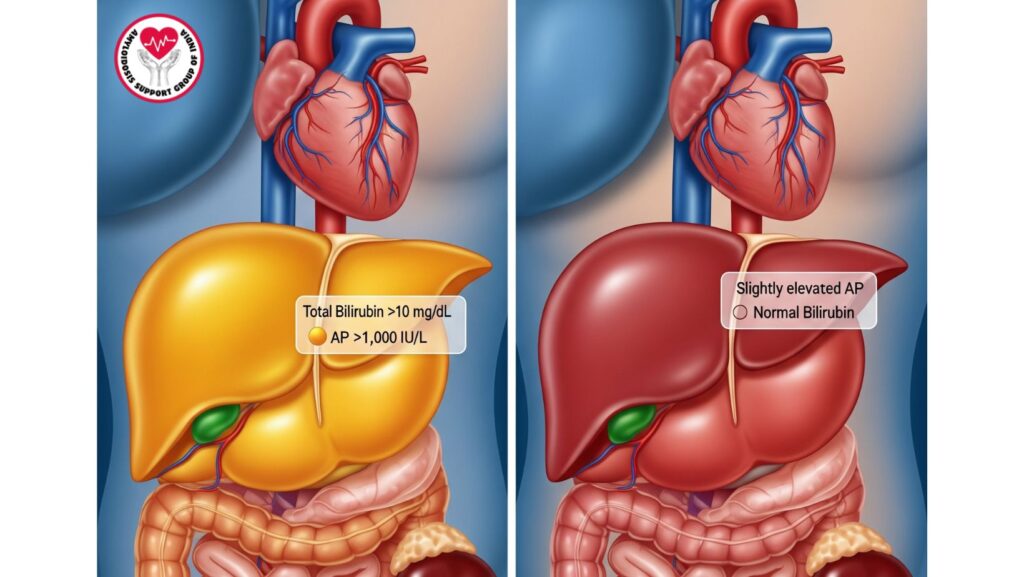
Table of Contents
Introduction
Systemic primary (AL) amyloidosis is a rare multisystem disorder due to misfolded immunoglobulin light chains that deposit as amyloid fibrils. Multiple organs, including the liver, heart, kidneys, and gastrointestinal tract, are involved. Hepatic involvement is present in 60–90% of patients, but the majority are subclinical, with minor liver function test (LFT) abnormalities or hepatomegaly.
One of the most important prognostic indicators is jaundice, indicative of extensive cholestatic liver disease. Patients with jaundice have a median survival of about 3 months, while those with liver disease but no jaundice have a much longer survival, with a median of around 2 years. These variations are critical for clinicians to use in diagnosis, treatment, monitoring, and palliative care.
This article discusses:
- Epidemiology and prevalence of liver involvement in AL amyloidosis
- Clinical presentation of patients with and without jaundice
- Laboratory and imaging results
- Prognosis and survival comparison
- Management approach in both patient groups
Epidemiology
Prevalence of Liver Involvement
- Involvement of the liver is frequent, presenting in 60–90% of systemic AL amyloidosis patients.
- Although common involvement is seen, severe cholestatic liver disease is uncommon, in ~5–10% of patients.
Risk Factors for Severe Hepatic Disease
- Heavy systemic amyloid burden
- Light chain deposition at a fast rate
- Multi-organ involvement, especially cardiac and renal
- Male predominance has been seen in several case series
Pathophysiology
Amyloid Deposition in the Liver
- Sinusoids: Fibrils impair hepatocyte function, occasionally resulting in mild LFT derangement.
- Portal tracts: Bile duct compression results in cholestasis and jaundice.
- Vascular walls: Amyloid deposition may impair hepatic perfusion, worsening liver failure.
Mechanisms of Jaundice vs Non-Jaundice Presentation
- With jaundice: Marked bile duct obstruction, hepatocyte compression, and decreased synthetic function.
- Without jaundice: Minimal deposition; mild hepatomegaly, subtle LFT abnormalities, maintained liver function.
Clinical Features
Patients With Jaundice
- Marked jaundice – main presenting symptom
- Hepatomegaly, occasionally tender
- Pruritus because of cholestasis
- Ascites from portal hypertension and hypoalbuminemia
- Laboratory profile:
- Total bilirubin >10 mg/dL
- AP >1,000 IU/L
- Mild-moderate AST/ALT elevation
- INR prolonged because of diminished synthetic function
- Albumin decreased
Patients Without Jaundice
- Frequently asymptomatic; liver disease found incidentally
- Mild hepatomegaly
- Slight elevations in AP or AST/ALT
- Normal or mildly increased bilirubin
- Synthesis intact
Diagnostic Evaluation
Laboratory Assessment
- Liver function tests (LFTs): AP, AST, ALT, bilirubin, albumin, INR
- Serum protein electrophoresis (SPEP) and urine protein electrophoresis (UPEP)
- Serum free light chain assay to measure amyloidogenic proteins
Imaging
- Ultrasound: Hepatomegaly, echotexture change
- CT/MRI: Nodular liver in extreme cases, ascites, portal hypertension
- MRI Elastography: Assesses liver stiffness and fibrosis
Liver Biopsy
- Congo red staining: Diagnoses amyloid deposits
- Mass spectrometry: Diagnoses AL type
- Transjugular biopsy: Advised in coagulopathic or ascitic patients
Prognosis and Survival
Survival in Jaundiced Patients
- Median survival: ~3 months
- Sudden progression to hepatic synthetic failure
- Often comes with multi-organ involvement
- Complications are at high risk: ascites, coagulopathy, and systemic decompensation
Survival in Patients Without Jaundice
- Median survival: ~2 years
- Usual milder hepatic involvement
- Usually asymptomatic or found incidentally
- Prognosis is more based on cardiac or renal involvement than liver disease
Comparative Insights
| Feature | With Jaundice | Without Jaundice |
| ———————– | —————– | ———————— |
| Median Survival | ~3 months | ~2 years |
| Bilirubin | Markedly elevated | Normal/slightly elevated |
| AP | >1,000 IU/L | Mildly elevated |
| Synthetic Function | Often impaired | Usually preserved
| Multi-Organ Involvement | Common | Less frequent |
Management Strategies
Patients With Jaundice
- Plasma cell-directed therapy: Bortezomib, cyclophosphamide, dexamethasone, daratumumab
- Supportive care: Diuretics, nutritional support, pruritus management
- Advanced interventions: Occasionally, isolated liver transplant if no multi-organ disease
- Palliative care: Early integration important due to poor prognosis
Patients Without Jaundice
- Systemic therapy based on plasma cell disease and organ involvement
- Routine monitoring of liver function
- Supportive care as required for mild hepatic symptoms
- Long-term prognosis usually hinges on cardiac and renal amyloidosis
Case Studies
- Patient A: Severe jaundice, hepatomegaly, ascites; biopsy proved AL amyloidosis. Multi-organ involvement ruled out transplant; survival 3 months.
- Patient B: Mild hepatomegaly, minimal LFT abnormalities, no jaundice. Treated with chemotherapy; survival >2 years.
- Patient C: Incidental hepatic AL amyloidosis, asymptomatic, followed; responded to systemic therapy; survival >3 years.
Clinical Pearls
- Jaundice is sign of high-risk liver involvement in AL amyloidosis.
- Nonjaundiced patients can have indolent hepatic disease and significantly longer survival.
- Early diagnosis through biopsy is critical for prognosis and treatment.
- Multidisciplinary care enhances outcomes, especially in systemic disease.
- Palliative care is essential in patients with advanced cholestatic liver failure.
Future Directions
- New therapies aimed at hepatic amyloid deposits
- Creation of biomarkers for the early identification of high-risk patients
- Clinical trials investigating transplantation, combination regimens, and early treatment
- Imaging studies to monitor disease progression
Conclusion
Liver disease in AL amyloidosis shows a range from mild, subclinical illness to cholestatic liver failure with jaundice. Survival varies dramatically:
- With jaundice: Median survival ~3 months; frequently multi-organ disease, severe prognosis.
- Without jaundice: Median survival ~2 years; milder liver disease, prognosis dependent upon other organ systems.
Being aware of this spectrum enables clinicians to: - Risk stratify patients
- Tailor systemic therapy
- Arrange timely palliative care
- Maximize patient quality of life
Early identification, multidisciplinary treatment, and individualized therapy continue to be crucial for enhancing outcomes in both groups of patients.
