
Can Amyloid Deposits Spread Between Cells?
Table of Contents
Introduction
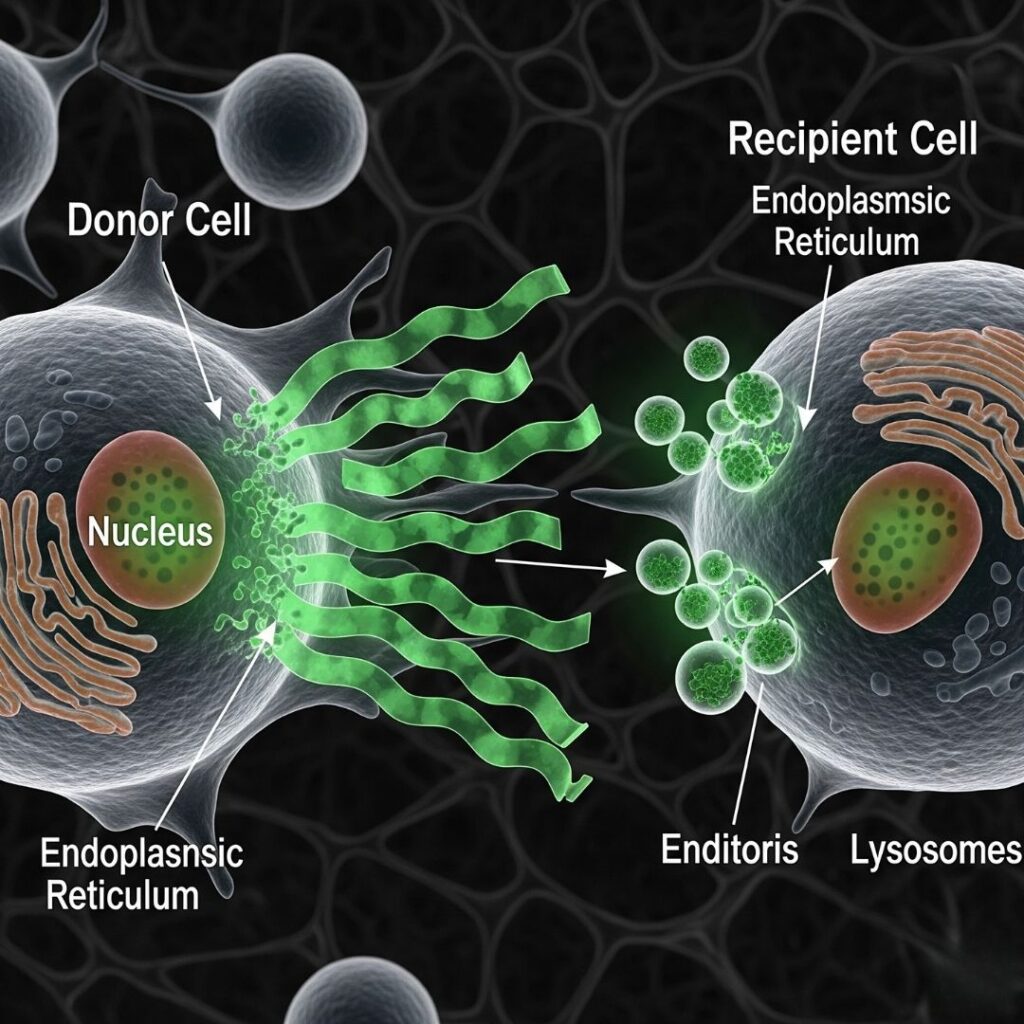
Amyloidosis is a multifactorial and multifunctional illness brought about by the deposition of misfolded protein aggregates, or amyloid fibrils, in organs and tissues. The protein deposits interfere with normal cellular processes and ultimately result in organ injury. Although once viewed as a localized phenomenon, there is new evidence to suggest that amyloidogenic proteins can have “prion-like” behavior—i.e., they can transmit their misfolded nature from cell to cell.
This finding is a paradigm shift in the way we think about amyloidosis. If amyloid fibrils are actually capable of spreading from one cell to another by templating additional aggregation, the disease progression, treatment, and prevention implications are enormous.
Throughout this in-depth blog post, we discuss the cell-to-cell transmission of amyloid fibrils, review the principle of prion-like mechanisms, and look at how this might change our practice regarding the treatment of amyloidosis.
1. Understanding Amyloidosis
Amyloidosis is a collection of diseases brought about by the extracellular deposition of insoluble protein fibrils. These amyloid fibrils are deposited in organs like the heart, kidneys, liver, and nervous system, causing structural and functional damage.
The most prevalent forms are:
- AL (Light Chain) Amyloidosis
- AA (Serum Amyloid A) Amyloidosis
- ATTR (Transthyretin) Amyloidosis
- Aβ Amyloidosis (Alzheimer’s disease)
In spite of variations in the protein precursors themselves, all these diseases have a unified thread: the pathologic deposition of misfolded proteins.
2. What are Amyloid Fibrils?
Amyloid fibrils are well-ordered, β-sheet-dense structures produced when proteins in their native conformation misfold and aggregate. These fibrils are insoluble and can be deposited in tissues over time.
Key Features:
- Insoluble in water
- Resistant to proteolysis
- Stain with Congo red dye (apple-green birefringence under polarized light)
- Display cross-β sheet X-ray diffraction patterns
Amyloid fibrils are not inert byproducts alone. They can bind to cellular machinery, immune receptors, and even replicate themselves—like prions.
3. The Idea of Protein Misfolding
Proteins are assembled as linear amino acid chains that need to fold into precise 3D structures to be functional. Environmental stress, mutations, or disruptions in proteostasis can trigger misfolding.
When misfolded proteins:
- Do not refold or degrade
- Oligomerize or fibrilize
- Perturb cellular function
- Trigger oxidative stress and inflammation
This theory underlies not just amyloidosis but also neurodegenerative disorders such as Alzheimer’s, Parkinson’s, and Huntington’s disease.
4. What Is “Prion-Like”?
The phrase “prion-like” comes from prion diseases (for example, Creutzfeldt–Jakob disease), in which a misfolded variant of the prion protein (PrP^Sc) causes normal prion proteins to misfold.
Prion-like behavior includes:
- Self-propagation of misfolded structure
- Cell-to-cell transmission
- Seeding new aggregates in recipient cells
In amyloidosis, that implies that a misfolded amyloid protein can serve as a template, folding other proteins into the same misfolded, aggregation-prone state.
5. Mechanisms of Amyloid Spread Between Cells
A. Exosome-Mediated Transfer
Cells secrete exosomes with misfolded proteins that fuse with other cells and transfer their load.
B. Tunneling Nanotubes
Direct cell-to-cell contacts could permit the transfer of aggregated proteins from one cell to another.
C. Endocytosis and Phagocytosis
Amyloid fibrils secreted into the extracellular space could be engulfed by neighboring cells.
D. Seeding and Nucleation
Misfolded proteins are nucleation sites, which trigger the aggregation of native proteins around them.
6. Examples of Prion-Like Spread in Amyloid Diseases
A. Alzheimer’s Disease (Aβ and Tau proteins)
Aβ plaques and Tau tangles have demonstrated capacity to propagate through neural circuits.
B. Parkinson’s Disease (α-synuclein)
Misfolded α-synuclein spreads from the gut to the brain.
C. Systemic Amyloidosis (AL, AA, ATTR)
Although less well studied, recent evidence indicates systemic amyloid proteins might also propagate along similar routes.
7. Experimental Evidence Supporting Intercellular Spread
Animal Models:
- Injection of amyloid fibrils into transgenic mice accelerates amyloid formation in remote tissues.
- In models of Alzheimer’s, injection of fibrils of Aβ into the brain elicits plaque formation.
In Vitro Studies:
- Cells grown in culture and treated with misfolded proteins start producing their own fibrils.
- Fibril intercellular transmission has been seen by transfection of culture media shared between cells.
Human Studies:
- Iatrogenic transmission (contaminated surgical equipment) has been linked to limited instances of cerebral amyloidosis.
8. Effects of Amyloid Propagation
The transmission of misfolded proteins worsens:
- Tissue Damage: By cumulative amyloid burden.
- Organ Dysfunction: Especially in the heart, kidneys, and nerves.
- Disease Progression: Is hastened by systemic involvement.
- Therapeutic Resistance: Spread can interfere with treatment effectiveness.
Amyloid spread enhances severity of disease and can account for why progressively worse symptoms occur despite an initial localized course.
9. Immune System Involvement
The immune system can inadvertently augment amyloid spread.
- TLRs (Toll-Like Receptors) like TLR2 and TLR4 recognize amyloid fibrils and trigger inflammatory pathways (e.g., NF-κB).
- Microglia and macrophages take in amyloid, potentially spreading aggregates further.
- Chronic inflammation compromises tissues to misfolded protein deposition.
This inflammatory environment is a positive feedback loop, promoting additional misfolding and cellular stress.
10. Therapeutic Implications and Targeted Strategies
If amyloid spreading is prion-like, intervention strategies will need to do more than decrease production—algorithms must cut off propagation.
A. Immunotherapy
Monoclonal antibodies (e.g., patisiran, tafamidis) can neutralize extracellular amyloid seeds.
B. Seeding Inhibitors
Small molecules that inhibit the capacity of misfolded proteins to template subsequent aggregation.
C. Proteostasis Regulators
Increasing the cell’s capacity to break down misfolded proteins (e.g., through autophagy or proteasome activation).
D. Cellular Entry Inhibitors
Prevent internalization of extracellular fibrils by inhibiting uptake pathways.
E. Gene Editing Strategies
Amyloidogenic precursor gene silencing or editing (e.g., CRISPR-Cas9 therapies).
11. Future Directions
The concept of intercellular transmission of amyloid continues to evolve. Future studies need to address:
- Identifying the precise mechanisms of spread in systemic amyloidosis.
- Targeting specific cell pathways involved in transmission.
- Developing diagnostic markers that identify early propagation.
- Discovering vaccines that inhibit seed formation.
Interdisciplinary strategies integrating neurology, immunology, and molecular biology will be crucial.
12. Conclusion
There is a consensus that it is likely to be a testable hypothesis that amyloid deposits do communicate between cells.
Misfolded proteins are capable of serving as templates inducing the misfolding and aggregation of properly folded proteins, hence propagating the disease in a prion-like fashion.
This revelation has reshaped our understanding of amyloidosis, providing new insights into its progression and offering novel therapeutic targets. Interventions that halt the seeding and spread of misfolded proteins may represent the next frontier in the fight against amyloidosis.
By continuing to explore the cell-to-cell transmission of amyloid fibrils, researchers inch closer to more effective treatments and ultimately, a cure.
13. Frequently Asked Questions (FAQs)
Q1. What is “prion-like” in the case of amyloidosis?
It is the mechanism by which misfolded amyloid proteins are able to transmit their misfolded structure to regular proteins, as prions do.
Q2. Is amyloidosis infectious like prion diseases?
No. Amyloidosis is not contagious from person to person under normal circumstances. Under some laboratory or unusual medical conditions, seeding of proteins theoretically might occur.
Q3. Will stopping amyloid propagation enhance treatment?
Yes. Stopping the propagation could decelerate disease advancement and inhibit organ damage.
Q4. How do scientists identify cell-to-cell propagation?
By means of animal models, cell cultures, and biomarker assays quantifying the transfer and templating of misfolded proteins.
Q5. Are therapies on the horizon that intervene in this propagation?
Yes. Monoclonal antibodies, seeding inhibitors, and proteostasis enhancers are under active development and exploration.
