
Liver Failure in AL Amyloidosis – Rare but Universally Lethal Presentation
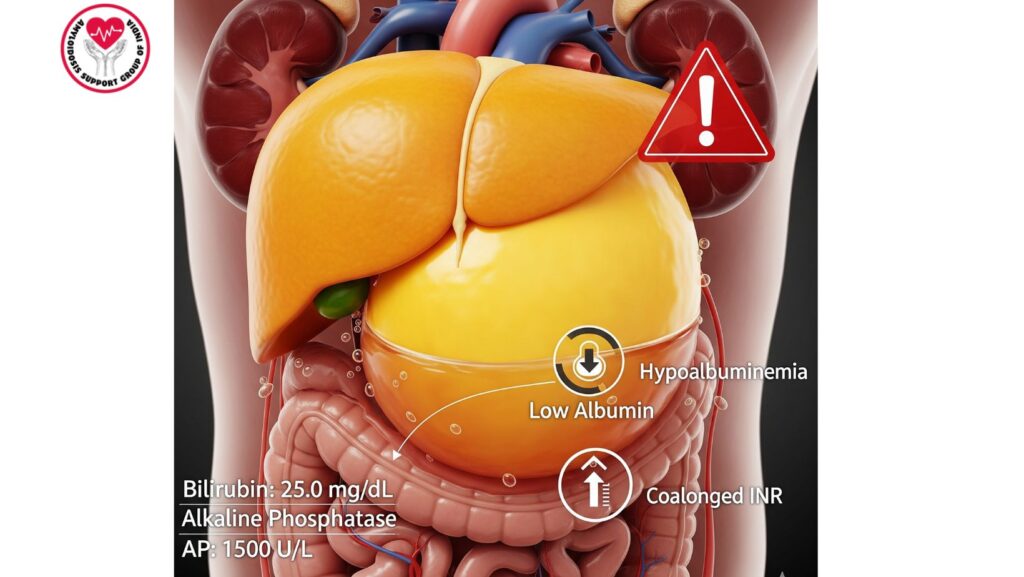
Table of Contents
Introduction
Systemic primary (AL) amyloidosis is a multisystem disorder of unusual frequency occurring due to the deposition of fibrils of misfolded immunoglobulin light chains as amyloid. These fibrils deposit in various organs such as the heart, kidneys, gastrointestinal tract, and liver.
Whereas liver involvement is present in 60–90% of patients with AL amyloidosis, severe liver failure is extremely uncommon. However, when it does happen, it is always fatal, underlining the urgent need for prompt recognition, vigorous supportive therapy, and judicious prognostication.
This article presents an in-depth review of:
- Epidemiology of liver involvement in AL amyloidosis
- Pathophysiology of severe hepatic amyloid deposition
- Clinical features of liver failure in this disease
- Diagnostic strategies
- Prognostic significance
- Management problems and supportive care
- Directions for research and therapy in the future
Epidemiology
Liver Involvement Prevalence
- Liver involvement is seen in 60–90% of systemic AL amyloidosis, predominantly subclinical.
- Clinically evident liver disease, including cholestatic liver failure, is extremely uncommon, seen in only about 5–10% of cases with involvement of the liver.
Demographics
- Affects usually older adults, typically between 55–70 years.
- No predominant gender, although some case series report minor male excess.
Risk Factors for Severe Hepatic Disease
- Heavy systemic amyloid burden
- Amyloidogenic light chain deposition at an accelerated rate
- Multi-organ involvement, especially cardiac and renal
- Certain light chain subtypes with high amyloidogenicity
Pathophysiology of Liver Failure in AL Amyloidosis
Mechanisms of Hepatic Dysfunction
- Sinusoidal amyloid deposition: Compresses hepatocytes, interfering with bile flow.
- Portal tract infiltration: Causes cholestasis and defective bile excretion.
- Vascular compromise: Decreased hepatic perfusion aggravates hepatocellular damage.
- Combined multi-organ dysfunction: Aggravates metabolic disturbances and systemic manifestations.
Consequences of Hepatic Amyloidosis
- Cholestasis: Results in jaundice, pruritus, and bilirubin accumulation.
- Synthetic dysfunction: Hypoalbuminemia, coagulopathy, and ascites.
- Metabolic impairment: Restricts the use of conventional chemotherapeutic agents metabolized by the liver.
Clinical Features
Presentation
Patients with liver failure from AL amyloidosis usually have:
- Jaundice: Sickle and quickly progressive
- Hepatomegaly: Occasionally tender
- Pruritus: Due to cholestasis
- Ascites: Secondary to hypoalbuminemia and portal hypertension
- Fatigue and malaise: Frequent systemic symptoms
Laboratory Findings
- Total bilirubin: Often very elevated (>10 mg/dL)
- Alkaline phosphatase (AP): Often >1,000 IU/L
- AST/ALT: Mild to moderate elevation
- Albumin: Low from impaired synthesis or associated nephrotic syndrome
- INR: Elevated, indicating hepatic synthetic failure
Systemic Manifestations
- Multi-organ involvement is characteristic:
- Cardiac: Arrhythmias, heart failure
- Renal: Proteinuria, progressive renal failure
- Gastrointestinal: Malabsorption, nausea, anorexia
Diagnostic Approach
Laboratory Assessment
- Complete liver function panel: AST, ALT, AP, bilirubin, albumin, INR
- Serum and urine protein electrophoresis (SPEP/UPEP)
- Serum free light chain assay
Imaging
- Ultrasound: Hepatomegaly with heterogeneous echotexture
- CT/MRI: Nodular liver, ascites, portal hypertension
- MRI Elastography: Measures liver stiffness and fibrosis
Liver Biopsy
- Congo red staining: Diagnoses amyloid deposition
- Mass spectrometry: Diagnoses AL type
- Transjugular biopsy: Used in patients with coagulopathy or ascites
Prognosis
- Universal fatality: After liver failure occurs, prognosis is very poor
- Median survival: About 3 months after onset of jaundice
- Predictors of poor outcome:
- Total bilirubin >10 mg/dL
- AP >1,000 IU/L
- Rapid hepatic synthetic decline
- Involvement of multiple organs
Comparison of Survival
| Attribute | Liver Failure (Jaundice) | Liver Involvement Without Failure |
| ———————– | ———————— | ——————————— |
| Median Survival | ~3 months | ~2 years |
| Bilirubin | Significantly elevated | Mildly/slightly elevated |
| AP | >1,000 IU/L | Mildly elevated |
| Synthetic Function | Impaired | Typically preserved |
| Multi-Organ Involvement | Common | Less frequent |
Management Challenges
Limitations of Standard Therapy
- Bortezomib and melphalan: Metabolized in the liver; failure of the liver enhances the risk of toxicity.
- Cyclophosphamide: Dosage adjustment required in hepatic impairment.
- Monoclonal antibodies (daratumumab): Generally safer but cautious use may be needed.
- Severe hepatic impairment usually precludes vigorous chemotherapy.
Supportive Care
- Nutritional support of hypoalbuminemia
- Diuretics and paracentesis for ascites
- Symptom control: Pruritus, fatigue, anorexia
Palliative Care
- Early integration is recommended because of rapid progression and poor prognosis
- Emphasis on quality of life, symptom relief, and comfort
Liver Transplantation
- Not usually an option
- Usually contraindicated because of multi-organ involvement
- Only contemplated in isolated hepatic amyloidosis, which is very rare
Case Studies
- Patient A: Sudden onset jaundice, ascites, coagulopathy; biopsy diagnosed AL amyloidosis; expired 4 weeks after presentation.
- Patient B: Multi-organ AL amyloidosis with severe cholestatic liver failure; comfort care emphasized; survival 3 months.
- Patient C: Isolated hepatic amyloidosis; underwent transplant; patient survived >2 years.
Clinical Pearls
- Liver failure in AL amyloidosis is rare but always fatal.
- Early identification of hepatic amyloidosis is key to monitoring and supportive therapy.
- Intensive chemotherapy could be contraindicated because of hepatic metabolism constraints.
- Multidisciplinary and palliative strategies are required for patient-centered therapy.
- Prognosis is generally based on degree of hepatic and multi-organ involvement.
Future Directions
- Development of hepatic-sparing therapies
- Clinical trials of new drugs in patients with extensive liver involvement
- Biomarkers for early identification of high-risk hepatic amyloidosis
- Imaging for disease monitoring
Conclusion
Liver involvement is prevalent in AL amyloidosis, but liver failure is exceptional but nearly always fatal. Key points:
- Severe cholestatic liver disease is a prognostic marker of gravity.
- Median survival from onset of liver failure is ~3 months.
- Treatment choices are limited by impaired hepatic metabolism.
- Supportive care and palliative measures dominate the management strategy.
- Multidisciplinary management is key to maximizing quality of life in this high-risk group.
Early recognition, prompt intervention, and attentive management are necessary to overcome the therapeutic and prognostic difficulties of liver failure in AL amyloidosis.
