
Severe Liver Manifestations in AL Amyloidosis – Understanding Cholestatic Liver Failure Subsets
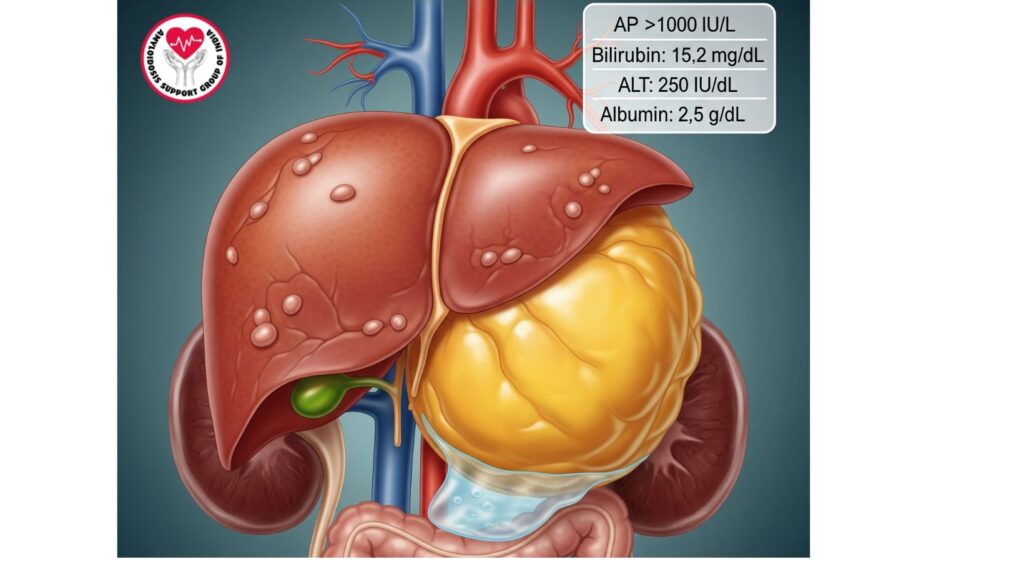
Table of Contents
Introduction
Systemic primary (AL) amyloidosis is a rare and frequently disabling illness due to immunoglobulin light chain misfolding, leading to amyloid fibril deposition in various organs. Although liver involvement affects 60–90% of patients, most have mild, clinically silent disease with minimal liver function test (LFT) abnormalities and hepatomegaly.
A subset of patients, but with severe cholestatic liver failure, with pronounced jaundice, pruritus, coagulopathy, and rapid evolution into liver dysfunction. This population is a high-risk group with substantial morbidity and mortality and requires early identification, proper diagnosis, and cautious management.
This article provides a comprehensive review of severe liver manifestations of AL amyloidosis, with emphasis on pathophysiology, clinical presentation, diagnostic methods, management, and prognostication.
Epidemiology
Severity of Severe Hepatic AL Amyloidosis
- Although 60–90% of AL amyloidosis patients have liver disease, only a minority (~5–10%) are severely cholestatic liver failures.
- Patients are usually elderly, with mean age of 60 years at presentation.
- Slight male predominance is noted in several case series.
Risk Factors
- Widespread systemic amyloid deposition (multi-organ)
- Rapid disease progression of AL amyloidosis
- Underlying plasma cell dyscrasias with highly amyloidogenic light chains
Pathophysiology
Severe liver disease in AL amyloidosis results from massive amyloid deposition in hepatocytes, sinusoids, and biliary tracts:
- Sinusoidal deposition – interferes with normal hepatocyte function and blood flow.
- Portal tract infiltration – causes compression of bile ducts, resulting in cholestasis.
- Vascular involvement – impairs hepatic perfusion, leading to ischemia and hepatic insufficiency.
Mechanisms of Cholestatic Liver Failure
- Bile flow obstruction due to amyloid deposits surrounding bile canaliculi and ducts.
- Hepatocellular damage due to progressive sinusoidal compression.
- Combined organ dysfunction worsens hepatic failure (e.g., renal or cardiac involvement causing volume overload).
Clinical Features
Those with severe cholestatic liver failure present with the following:
Hepatic Signs
- Severe hepatomegaly
- Jaundice – frequently severe and rapidly developing
- Pruritus – secondary to cholestasis
- Ascites – secondary to portal hypertension and hypoalbuminemia
- Tenderness – can occur in advanced disease
Laboratory Findings
- Total bilirubin: Usually >10 mg/dL
- Alkaline phosphatase (AP): Significantly increased, often >1,000 IU/L
- AST/ALT: Mild-to-moderate elevation
- INR: Can be prolonged due to altered synthetic function
- Albumin: Reduced in association with nephrotic syndrome or liver synthetic dysfunction
Systemic Manifestations
- Fatigue and anorexia
- Edema – lower extremity or generalized
- Signs of multi-organ involvement – cardiac arrhythmias, renal failure
Diagnostic Evaluation
Laboratory Evaluation
- Liver function tests (LFTs) – increased AP, bilirubin, mild transaminase elevation
—|—
- Coagulation profile – measures synthetic activity
- Serum protein electrophoresis (SPEP) – identifies monoclonal light chains
- Serum free light chain assay – measures amyloidogenic proteins
Imaging
—|—
- Ultrasound: Hepatomegaly, inhomogeneous echotexture
- CT scan: Nodular liver, ascites; can identify portal hypertension
- MRI: Can characterize infiltrative pattern and exclude other hepatic disease
Liver Biopsy
- Definitive diagnosis – Congo red staining reveals apple-green birefringence
- Mass spectrometry – confirms AL type
- Transjugular biopsy – considered in coagulopathy or ascites
Prognostic Implications
- Mortality is high for severe cholestatic liver failure in AL amyloidosis, with median survival frequently <6 months from jaundice onset.
- Prognostic markers:
- Total bilirubin >10 mg/dL
- AP >1,000 IU/L
- Rapid worsening of liver synthetic function
- Multi-organ involvement severely compromises prognosis; isolated hepatic disease is uncommon
Strategies for Managing the Disease
Medical Treatment
- Plasma cell-directed therapy: Bortezomib, cyclophosphamide, dexamethasone, daratumumab
- Supportive care:
- Diuretics for ascites
- Dietary support
- Symptom management for pruritus and fatigue
Intensive Therapies
- Liver transplantation: Rarely considered in isolated hepatic involvement, not usually possible in multi-organ disease
- Combined organ transplantation: Reserved for highly selected patients with severe systemic disease
Palliative Care
- In rapidly progressive multi-organ-involvement patients, conversion to comfort care can be indicated
- Emphasis on quality of life, symptom control, and dignity
Case Studies and Literature Review
- Case 1: Patient had severe jaundice, hepatomegaly, and elevated AP. Biopsy was consistent with AL amyloidosis. Multi-organ involvement made transplantation impossible; patient treated with chemotherapy and supportive care, survival 3 months.
- Case 2: Sudden cholestatic liver failure with AL amyloidosis and renal and cardiac involvement. Hospice care transitioned; died within 4 weeks.
- Case 3: Isolated hepatic AL amyloidosis; successful liver transplant with systemic therapy; survived >2 years.
Clinical Pearls
- High index of suspicion necessary in AL amyloidosis patients presenting with jaundice and hepatomegaly.
- Early diagnosis and biopsy are paramount to ensure proper therapy and prognostication.
- Systemic therapy may stabilize organ function in carefully selected patients but only infrequently reverses profound cholestatic liver failure.
- Integration of palliative care is mandatory in end-stage, multi-organ disease.
Future Directions
- New therapies for amyloid deposits within the liver
- Biomarkers for early identification of high-risk patients
- Clinical trials investigating transplantation and combination therapy in isolated hepatic AL amyloidosis
- Advanced imaging modalities to track progression of disease
Conclusion
Severe cholestatic liver failure is a high-risk subset of AL amyloidosis patients, having rapid disease progression and poor prognosis. Early diagnosis, precise diagnosis, and multidisciplinary care are crucial.
Key points:
- The majority of AL amyloidosis liver disease is mild, but severe cholestatic liver failure is uncommon but disastrous.
- Severe jaundice, hepatomegaly, marked elevation of AP, and multi-organ involvement denote poor prognosis.
- Management emphasizes systemic therapy, supportive care, and palliative interventions.
Knowledge of this subset enables clinicians to offer timely interventions, optimize patient care, and plan end-of-life strategies effectively.
