
Table of Contents
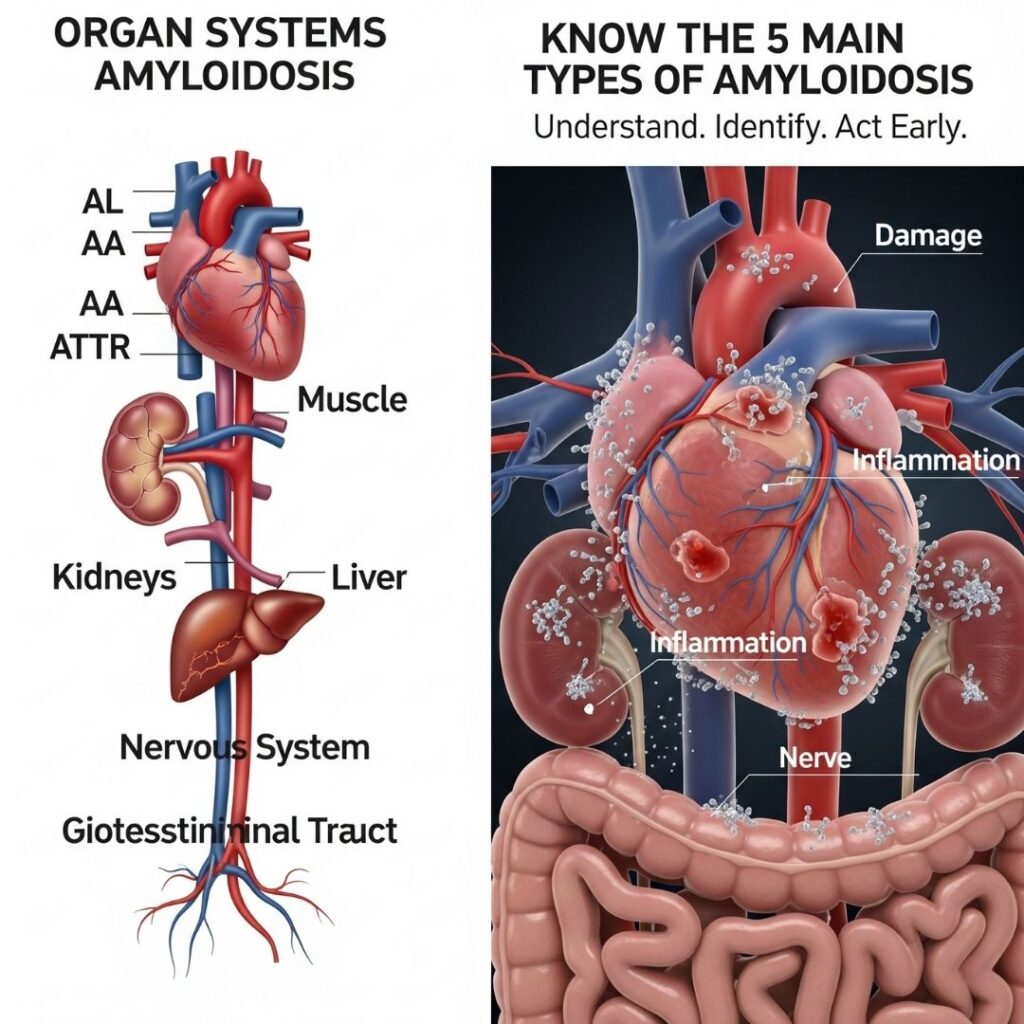
Introduction-types of amyloidosis
Amyloidosis is an uncommon but life-threatening disorder resulting from the aberrant deposition of amyloid proteins in organs and tissues. The misfolded proteins disrupt normal organ function and can cause potentially fatal complications. Although amyloidosis is not a disease itself, it describes a collection of diseases that vary in etiology, therapy, and outcome. Knowledge of the amyloidosis types is essential for timely diagnosis, successful treatment, and effective management. In this article, the author discusses the significant types: AL (Primary), AA (Secondary), Hereditary ATTR, Wild-type ATTR, and Dialysis-related amyloidosis.
1. AL Amyloidosis (Primary Amyloidosis)
Overview:
AL amyloidosis, also known as light chain amyloidosis, is the most prevalent type of the disease. It is linked with plasma cell disorders like multiple myeloma. In this condition, the bone marrow secretes faulty antibodies (light chains) that misfold and deposit as amyloid.
Causes:
- Plasma cell dyscrasias
- Multiple myeloma
Affected Organs:
- Kidneys (resulting in proteinuria)
- Heart (resulting in restrictive cardiomyopathy)
- Liver
- Nervous system
Symptoms:
- Fatigue
- Leg and ankle swelling
- Irregular heartbeat
- Shortness of breath
- Numbness or tingling in hands and feet
Diagnosis:
- Serum and urine protein electrophoresis
- Immunofixation
- Bone marrow biopsy
Treatment:
- Chemotherapy (same as treatments for multiple myeloma)
- Autologous stem cell transplant
2. AA Amyloidosis (Secondary Amyloidosis)
Overview:
AA amyloidosis is usually a complication of long-term inflammatory or infectious disease. It occurs because of deposition of Serum Amyloid A (SAA), an acute-phase protein
Causes:
- Rheumatoid arthritis
- Inflammatory bowel disease
- Chronic infections (e.g., tuberculosis)
Affected Organs:
- Kidneys
- Liver
- Spleen
Symptoms:
- Protein in urine (proteinuria)
- Edema
- Weight loss
- Fatigue
Diagnosis:
- Biopsy with AA protein deposits
- Blood tests for inflammation (e.g., CRP, ESR)
Treatment:
- Treatment of the underlying inflammatory condition
- Colchicine (particularly in familial Mediterranean fever)
3. Hereditary Amyloidosis (Familial ATTR)
Overview:
Hereditary amyloidosis results from mutations in genes encoding for specific proteins, the most common being transthyretin (TTR). These are inherited through families.
Common Mutations:
- TTR gene (most prevalent)
Inheritance Pattern:
- Autosomal dominant
Affected Organs:
- Heart
- Peripheral nerves
- Eyes
- Kidneys
Symptoms:
- Carpal tunnel syndrome
- Peripheral neuropathy
- Gastrointestinal symptoms
- Heart failure
Diagnosis:
- Genetic testing
- Tissue biopsy
Treatment:
- Liver transplant (to discontinue production of mutated TTR)
- TTR stabilizers (e.g., tafamidis)
- Gene-silencing therapies (e.g., patisiran, inotersen)
4. Wild-Type ATTR (Senile Systemic Amyloidosis)
Overview:
Wild-type ATTR amyloidosis, previously referred to as senile systemic amyloidosis, results from deposition of normal (non-mutated) transthyretin protein, mainly occurring in elderly men.
Causes:
- Changes in TTR structure with age
Affected Organs:
- Heart (causing restrictive cardiomyopathy)
Symptoms:
- Fatigue
- Breathlessness
- Leg swelling
- Abnormal heart rhythms
Diagnosis:
- Cardiac MRI
- Nuclear imaging (e.g., bone scintigraphy)
- Biopsy of affected tissue
Treatment:
- TTR stabilizers (tafamidis)
- Supportive cardiac treatment
5. Dialysis-Related Amyloidosis (DRA)
Overview:
Dialysis-related amyloidosis is a condition found in individuals who are receiving long-term dialysis. It is brought about by the deposition of beta-2 microglobulin, a protein which is not efficiently removed by dialysis
Causes:
- Chronic hemodialysis (typically >5 years)
Organs and Structures Involved:
- Joints
- Bones
- Tendons
Symptoms:
- Stiffness and pain of the joints
- Bone cysts
- Carpal tunnel syndrome
Diagnosis:
- X-ray evidence of bone cysts
- Tissue biopsy of affected tissue
Treatment:
- Conversion to high-flux dialysis membranes
- Kidney transplant
Diagnosis and Detection of Amyloidosis
Although symptoms may be greatly varied based on type and involved organs, early detection is essential. Diagnostic technologies include:
- Blood tests and urine tests
- Tissue biopsies (abdominal fat pad, rectal, or organ-specific)
- Genetic testing (for familial types)
- Imaging studies (e.g., echocardiogram, MRI, nuclear scans)
Treatment Overview by Type
| Type | Primary Treatment | Additional Measures |
|---|---|---|
| AL Amyloidosis | Chemotherapy, stem cell transplant | Monitoring of heart and kidney function |
| AA Amyloidosis | Management of inflammatory disease | Nutrition support |
| Hereditary ATTR | Liver transplantation, gene therapy | Genetic counseling |
| Wild-type ATTR | Tafamidis, cardiac treatment | Lifestyle changes |
| Dialysis-Related | Kidney transplant | Pain and orthopedic management |
Living with Amyloidosis
Life with amyloidosis involves continued medical care, emotional well-being, and patient education. Support group membership, knowledge of advancements in treatment, and routine check-ups are crucial for maintaining long-term health.
Supportive Resources:
- Patient communities (e.g., amyloidosissupport forums)
- Genetic counseling services
- Specialized amyloidosis centers
Conclusion
Amyloidosis is a multifaceted and diverse disease with several types, each having its causes, symptoms, and therapies. Awareness and early detection are crucial in the management of this orphan disease. Whether AL, AA, hereditary ATTR, wild-type ATTR, or dialysis-related, the distinctions assist patients and carers in taking intelligent choices. If you or someone you know is displaying unexplained signs or has a family history of amyloidosis, see a healthcare specialist and have genetic testing.
Stay informed. Get tested. Empower your journey.
